«`html
Protein Annotation-Improved Representations (PAIR): A Flexible Fine-Tuning Framework that Employs a Text Decoder to Guide the Fine-Tuning Process of the Encoder
Языковые модели белков (PLM) обучаются на больших белковых базах данных для прогнозирования последовательностей аминокислот и генерации векторов признаков, представляющих белки. Эти модели доказали свою полезность в различных приложениях, таких как прогнозирование складывания белков и эффектов мутаций. Одной из ключевых причин их успеха является способность захватывать консервативные последовательные мотивы, которые часто важны для фитнеса белков. Однако эволюционные и окружающие факторы могут влиять на отношение между сохранением последовательности и фитнесом, что делает его сложным. PLM полагаются на псевдо-вероятностные цели, но включение дополнительных источников данных, таких как текстовые аннотации, описывающие функции и структуры белков, может улучшить их точность.
Исследователи из Университета Торонто и Института Вектор провели исследование, которое улучшило PLM, настраивая их с помощью текстовых аннотаций из UniProt, сосредотачиваясь на девятнадцати типах экспертно подготовленных данных. Они представили фреймворк Protein Annotation-Improved Representations (PAIR), который использует текстовый декодер для направления обучения модели. PAIR значительно улучшил производительность моделей в задачах прогнозирования функций, даже превзойдя алгоритм поиска BLAST, особенно для белков с низкой схожестью последовательностей с обучающими данными. Этот подход подчеркивает потенциал интеграции разнообразных текстовых аннотаций для продвижения обучения представлений белков.
Область маркировки белков традиционно полагается на методы, такие как BLAST, который обнаруживает гомологию последовательностей белков через выравнивание последовательностей, и скрытые модели Маркова (HMM), которые включают дополнительные данные, такие как семейство белков и эволюционную информацию. Эти классические подходы хорошо справляются с последовательностями с высокой схожестью, но испытывают трудности с обнаружением отдаленной гомологии. Эта проблема привела к разработке PLM, которые применяют техники глубокого обучения для изучения представлений белков из масштабных данных последовательностей, вдохновленных моделями обработки естественного языка. Недавние достижения также интегрируют текстовые аннотации, с моделями, такими как ProtST, использующие разнообразные источники данных для улучшения прогнозирования функций белков.
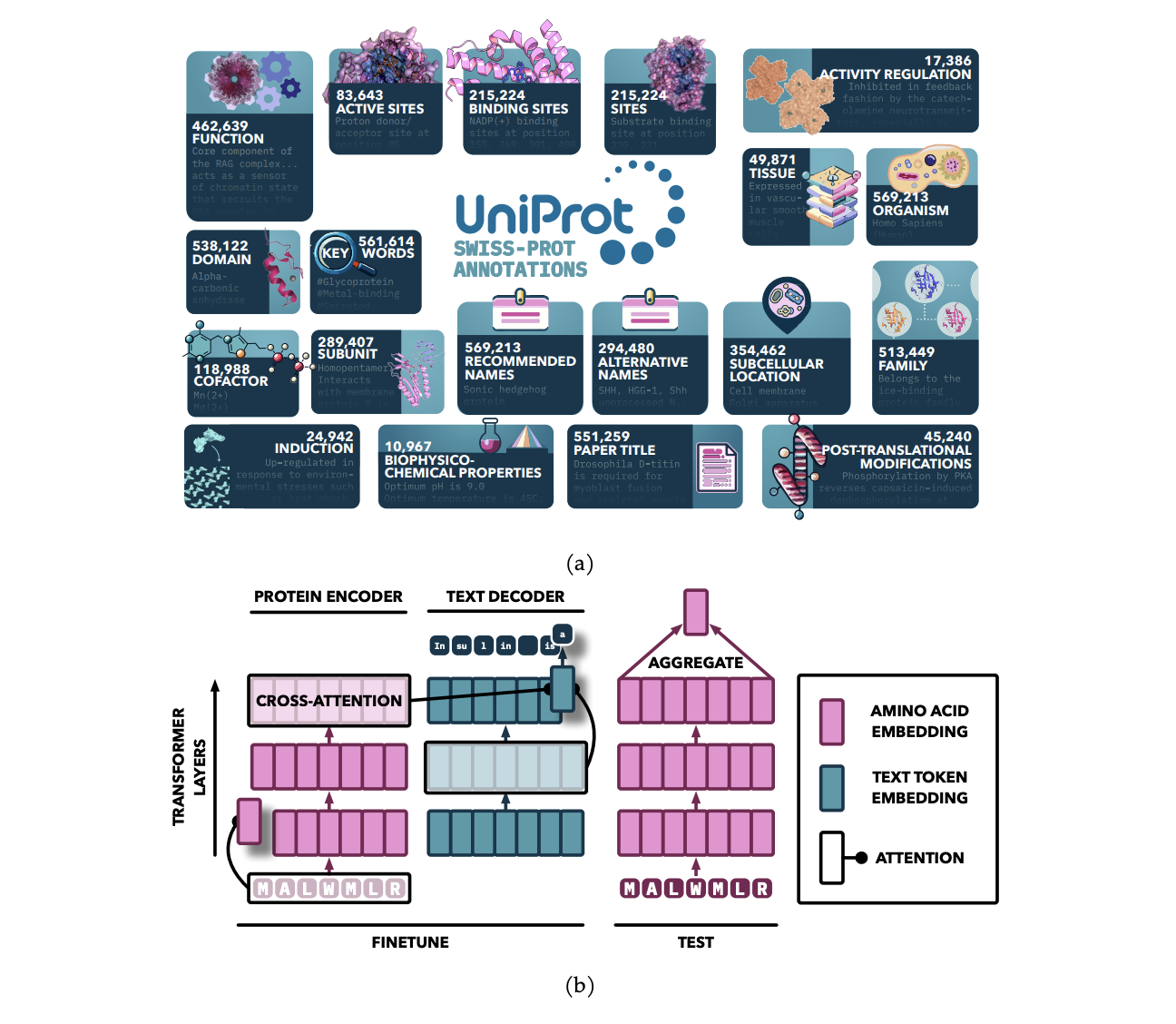
Модель использует архитектуру последовательности-к-последовательности на основе внимания, инициализированную предварительно обученными моделями и улучшенную добавлением кросс-внимания между кодировщиком и декодером. Кодировщик обрабатывает последовательности белков в непрерывные представления с использованием самовнимания, в то время как декодер генерирует текстовые аннотации авторегрессивным образом. Предварительно обученные модели белков из семейств ProtT5 и ESM служат в качестве кодировщика, в то время как SciBERT является текстовым декодером. Модель обучается на нескольких типах аннотаций с использованием специализированного подхода к выборке, обучение проводится на кластере HPC с использованием многократного обучения с точностью bfloat16.
Фреймворк PAIR улучшает прогнозирование функций белков путем настройки предварительно обученных моделей трансформера, таких как ESM и ProtT5, на высококачественных аннотациях из баз данных, таких как Swiss-Prot. Интеграция кросс-внимания позволяет текстовым токенам обращаться к последовательностям аминокислот, улучшая отношение между последовательностями белков и их аннотациями. PAIR значительно превосходит традиционные методы, такие как BLAST, особенно для белков с низкой схожестью последовательностей, и проявляет сильную обобщаемость к новым задачам. Его способность обрабатывать сценарии с ограниченными данными делает его ценным инструментом в биоинформатике и прогнозировании функций белков.
Фреймворк PAIR улучшает представления белков, используя разнообразные текстовые аннотации, которые отражают важные функциональные свойства. Путем объединения этих аннотаций PAIR значительно улучшает прогнозирование различных функциональных свойств, включая те, которые ранее не были характеризованы. PAIR последовательно превосходит базовые языковые модели белков и традиционные методы, такие как BLAST, особенно для последовательностей с низкой схожестью с обучающими данными. Результаты свидетельствуют о том, что интеграция дополнительных модальностей данных, таких как 3D структурная информация или геномные данные, может обогатить представления белков. Гибкий дизайн PAIR также имеет потенциальные применения для представления других биологических объектов, таких как малые молекулы и нуклеиновые кислоты.
Проверьте статью и модель. Вся заслуга за это исследование принадлежит исследователям этого проекта.
«`