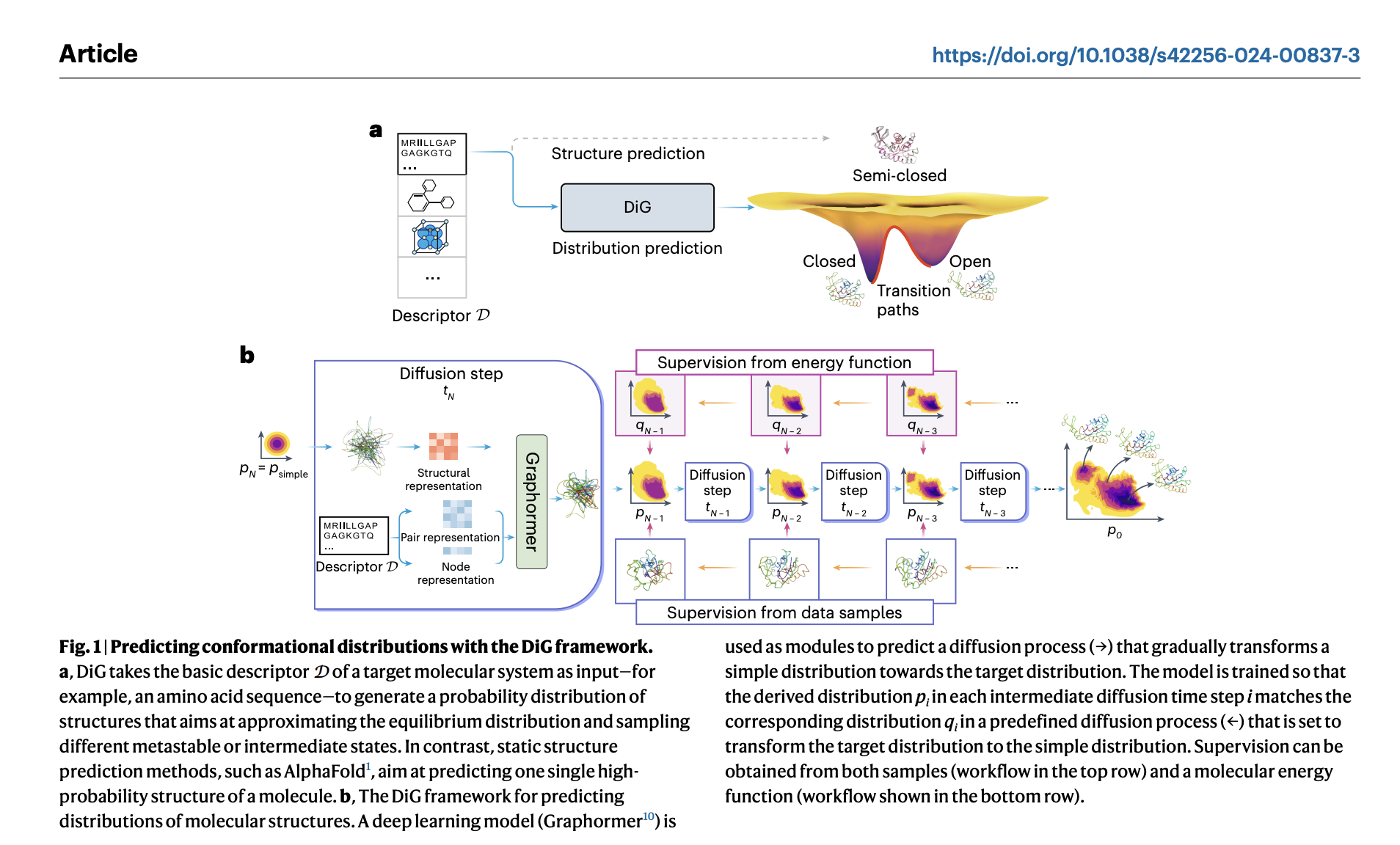
Прогресс в области глубокого обучения революционизировал предсказание структуры молекул, но в реальном мире часто требуется понимание равновесных распределений, а не только отдельных структур. Текущие методы, такие как молекулярно-динамические симуляции, являются вычислительно интенсивными и недостаточны для захвата полного спектра молекулярной гибкости. Предсказание равновесного распределения имеет ключевое значение для оценки макроскопических свойств и функциональных состояний молекул, таких как аденилаткиназа. Глубокое обучение показало перспективы в грубозернистых симуляциях, однако оно испытывает трудности с обобщением. Генераторы Больцмана предлагают потенциальное решение путем генерации равновесных распределений, однако их применимость к различным молекулам все еще нуждается в улучшении.
Исследователи из Microsoft Research AI4Science, Пекин, Китай; Университет науки и технологий Китая, Microsoft Quantum, Редмонд, Вашингтон, США; и Microsoft Research AI4Science, Берлин, Германия, разработали Distributional Graphormer (DiG), глубокую систему обучения, направленную на предсказание равновесного распределения молекулярных систем. Вдохновленный термодинамическим отжигом, DiG использует нейронные сети для преобразования простого распределения к равновесию на основе молекулярных дескрипторов, таких как химические графы или последовательности белков. Это позволяет эффективно генерировать разнообразные конформации и оценивать плотности состояний значительно быстрее, чем традиционные методы. DiG демонстрирует универсальность в различных молекулярных задачах и способен обобщаться на различные молекулярные системы. DiG приближает равновесное распределение путем симуляции процесса диффузии, облегчая предсказание молекулярных свойств и обеспечивая обратное проектирование структур с желаемыми свойствами.
Преимущества DiG:
- Преобразование равновесного распределения молекулярных систем с помощью глубокого обучения
- Генерация различных молекулярных структур с высокой скоростью
- Возможность обратного проектирования структур с желаемыми свойствами
- Универсальность и способность обобщаться на различные молекулярные системы
- Эффективное предсказание молекулярных свойств и плотностей состояний
- Точное воспроизведение сложных конформационных распределений
- Ускорение процесса открытия молекулярных структур и материалов
DiG также позволяет генерацию структур с учетом свойств и интерполяцию между состояниями, отображая структуры в латентное пространство. Этот инновационный подход совершенствует моделирование молекулярных структур, предлагая эффективное предсказание равновесных распределений и облегчая создание структур с учетом свойств.
Практические применения DiG:
- Генерация разнообразных структур белков для понимания их поведения и взаимодействий
- Точное предсказание структур лигандов в активных карманах для дизайна лекарств
- Эффективная идентификация активных мест на поверхности катализаторов
- Возможность обратного проектирования структур с желаемыми свойствами, такими как электронные зоны пропускания
В заключение, DiG революционизирует молекулярные науки, предсказывая равновесные распределения эффективно и обеспечивая разнообразную выборку молекул, важную для понимания взаимосвязи структуры и функции, а также для проектирования молекул и материалов. DiG обучается представлениям молекул по дескрипторам, таким как последовательности белков или формулы соединений, с помощью продвинутых архитектур глубокого обучения, точно описывая сложные распределения в многомерном пространстве. Его преимущество скорости перед традиционными методами, такими как молекулярно-динамические симуляции или сэмплирование методом MCMC, обладает трансформационным потенциалом, значительно снижая вычислительные затраты. Способность исследовать огромные конформационные пространства позволяет DiG ускорить открытие молекулярных структур, повлиять на различные области, включая науки о жизни, дизайн лекарств, катализ и науку о материалах.